مجموعه داده گرافی ESOL (میزان انحلال در آب)
مجموعه دادهی ESOL از داده هایی با ساختار گراف مناسب برای کار با شبکه های عصبی گرافی است.
این مجموعه داده شامل اطلاعات انحلالپذیری آب برای ۱۱۲۸ ترکیب شیمیایی است. این مجموعه داده برای آموزش مدلهایی استفاده شده است که به صورت مستقیم انحلالپذیری را از ساختار شیمیایی (بر اساس رشتههای SMILES) تخمین میزنند.
توجه داشته باشید که این ساختارها شامل مختصات سه بعدی نمیشوند، زیرا حلالیت ویژگی یک مولکول است و نه conformer آن. Conformers به شکلهای مختلفی گفته میشوند که یک مولکول میتواند در آنها وجود داشته باشد. این شکلها به دلیل چرخش حول پیوندهای شیمیایی و حرکت آزادانه برخی از اجزاء مولکول شکل میگیرند. مهم نیست که مولکول در کدام یک از conformerها قرار داشته باشد، خواص مولکول بر اساس ساختار کلی آن وابسته هستند و نه به شکل خاص آن. در نتیجه، ساختارهای المانی که برای توصیف محلول پذیری مولکول ها استفاده میشود عموماً شامل مختصات سه بعدی نیستند.
SMILES چیست؟
SMILES یک رشته کدگذاری شده است که ساختار شیمیایی یک مولکول را نشان میدهد. در توضیحات دیتاست در بالا، SMILES به منظور کدگذاری ساختار شیمیایی ترکیبات استفاده شده است تا بتوان با استفاده از آنها مدلهایی برای تخمین حلالیت مولکولی طراحی کرد.
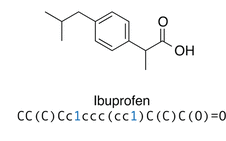
وظیفه یادگیری ماشین: چگونگی حل شدن مولکولهای مختلف در آب؟
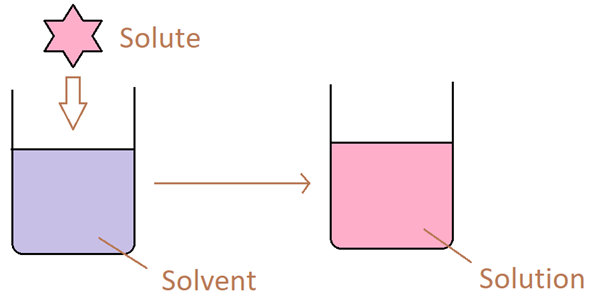
در شکل زیر، در سمت چپ، حلال (solvent) و حلشونده(solute) وجود دارند. حلال یک ماده است که قابلیت حل کردن دیگر مواد را دارد و به عنوان یک محیط برای حل کردن سایر مواد استفاده میشود. محلول(solution) نیز به مخلوطی از یک یا چند ماده در یک حلال گفته میشود که در شکل سمت راست نمایش داده شده است (مایع صورتی).
به عبارت دیگر، محلول، ترکیبی است که شامل حلال و حلشونده (ماده یا موادی که درون حلال حل شدهاند) میباشد.
حال این دیتاست در مورد میزان حل شوندگی مولکولهای شیمیایی مختلف در آب است که باید بتوانیم بر اساس مولکولهایی که قبلا آن ها و میزان حل شوندگی آنها در آب را دیدیم بتوانیم در مورد مولکولهایی که ندیدیم پیشبینی انجام بدیم. در واقع با یک مساله رگرسیون برای پیش بینی میزان انحلال هر مولکول مواجه هستیم. حال سوال آن است که چرا از خود رشته کدگذاری شدهی SMILES به عنوان ورودی مدل استفاده نکنیم و چرا ساختار گراف بهتر خواهد یود؟!
– استفاده از رشته SMILES ساده به عنوان ورودی مناسب نیست.
– استفاده از این رشته باعث میشود مدل به ساختار مولکول توجه نکند و به جای آن دستور زبان رشته SMILES را در نظر گرفته و به آن توجه کند.
– رشته SMILES برای یک مولکول ممکن است متفاوت باشد، بسته به نمایش (یک مولکول یکتا میتواند چندین رشته SMILES داشته باشد)
– اما گراف یک مادهی شیمیایی نسبت به جایگشتهایش ثابت است.
تمام این موارد انتخاب شبکههای عصبی گرافی را برای حل این مساله به یک انتخاب عقلانی و منطقی تبدیل میکند.
بصری سازی دیتاست مولکولی در پایتون – تبدیل SMILES به مولکول های RDKit
RDKit یک کتابخانه متنباز در پایتون است که برای کار با شیمی محاسباتی و تحلیل داده های شیمیایی طراحی شده است. این کتابخانه قابلیت ایجاد، خواندن و نوشتن فایلهای شیمیایی را دارد و علاوه بر این قابلیتها، ابزارهایی برای تبدیل ساختار مولکولی، تعیین صفات فیزیکوشیمیایی و محاسبه شاخصهای شیمیایی را نیز فراهم می کند.
در این دیتاست برای تبدیل رشتههای SMILES به مولکول و رسم آن از این ابزار استفاده میشود:
from rdkit import Chem from rdkit.Chem.Draw import IPythonConsole molecule = Chem.MolFromSmiles(data[0]["OCC3OC(OCC2OC(OC(C#N)c1ccccc1)C(O)C(O)C2O)C(O)C(O)C3O"]) molecule
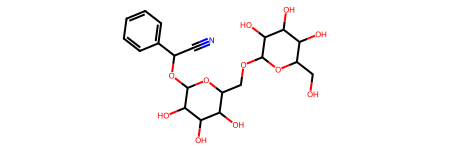
ما میتوانیم از این نمایش RDKit ویژگیهایی را برای گراف نهایی نیز بدست بیاوریم. در واقع این نوع نمایش به ما هر چیزی که نیاز داریم را بیان میکند، ویژگی هایی نظیر ویژگیهای اتم (نوع، … )، یالها، و غیره.
مطالب زیر را حتما مطالعه کنید

دیتاست کارت ملی ایرانی

مجموعه داده STL-10
مجموعه داده شناسایی حرکات دست

Persian Digits Audio Dataset

مجموعهدادهی The Stack

دیدگاهتان را بنویسید